Platelet Membrane Disorders and Impact-R
Wednesday, December 14th, 2011Inherited platelet disorders are rare conditions that are not usually encountered in clinical practice. However, the study of the pathophysiology has led to a better understanding of platelet biochemistry and physiology. They may be subdivided into the following:
- Platelet Membrane Disorders
- Platelet Granule Disorders
- Macrothrombocytopenias
- Platelet Signaling Disorders
The Platelet Membrane Disorders:
Glanzmann’s Thrombasthenia is a rare disorder where platelets can carry out biochemical reactions but are unable to form aggregates. This is an autosomal recessive trait where platelets have absent or dysfunctional GpIIb/IIIa complexes.
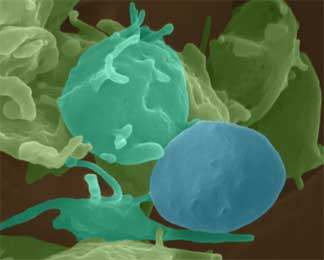
Smooth inactivated platelet (blue) with spiky activated platelets (light blue). ©2000 Dennis Kunkel, Ph.D.
In a normal platelet, there are about 50,000 of these complexes in the membrane. When platelets are activated, the complex binds fibrinogen, which in turn bind to GpIIb/IIIa complexes on other platelets, resulting in multicellular aggregates. Platelets are normal in size, shape and number. Patients with Glanzmann’s Thrombasthenia have mucosal bleeding throughout life and may even have severe bleeding episodes requiring platelet transfusions.
Bernard-Soulier Syndrome is another rare autosomal recessive disorder caused by mutations of the GpIb/IX/V complex. This complex is the main receptor for von Willebrand’s factor, which anchors platelets to exposed subendothelium in cases of endothelial injury and under high shear stress. The platelets are abnormally large and the count is low. Patients present with muco-cutaneous bleeding and prolonged bleeding time. They may require platelet transfusions and sometimes respond favourably to Desmopressin.
Pseudo- or Platelet type von Willebrand Disease is an autosomal dominant disorder arising from mutations of the GpIba polypeptide that make the platelet hypersensitive to vWF. Mucosal bleeding and borderline thrombocytopenia are noted. Pseudo-vWD resembles Type IIB vWD. An accurate diagnosis is necessary since treatment differs, Pseudo-vWD requiring platelet transfusions while Type IIB vWD requires vWF transfusion.
ADP receptor Deficiency is an autosomal recessive disorder involving the ADP receptor P2Y12. ADP released from damaged tissues and activated platelets plays a role in platelet aggregation through the mediation of receptors P2Y1 and P2Y12. P2Y1 initiates platelet response to ADP while P2Y12 forms and sustains large aggregates. Patients, therefore, have mild bleeding but are susceptible to posttraumatic and post-surgical blood loss.
Collagen receptor deficiency involves two receptors, an integrin protein, GpIa/IIa, and a non-integrin protein, GpVI. This deficiency is still under intense study.
Platelet function studies that show how platelets behave in vitro can be very helpful in diagnosis of these conditions. One of these is studying the platelet aggregates formed when blood is exposed to polystyrene in flow conditions. The IMPACT-R machine is one laboratory equipment that uses the cone and plate principal.